Basic Sciences: Bioinformatics
Diagnostic analysis of antibody profiles
Approximately 9 years ago, we started to develop algorithms for the diagnostic analysis of Western blots and electrophoretical separations.
We were the first to invent the use of multivariate analysis e.g., analysis of discriminance based on the digital image analysis of Western blots.
Additionally, we developed software modules to use different kind of
artitificial neural networks e.g., multi layer feedforward or probalistic neural networks for the diagnostic analysis of western blots.
Over the past years, we were able to prove the usefulness of our approaches in international cooperation projects.
Currently, we are developing algorithms to support our ongoing international cooperations for the analysis of autoantibody profiles in Johns Hopkins University (USA), University of Bern (Switzerland), and the University of Essen (Germany).
Development of new algorithms for proteomics analysis
One of the greatest challenges in proteomics is to handle and analyze the huge amount of protein data. For example, in a typical protein profiling study by means of Seldi-TOF proteinchips including pre-fractionation steps, several gigabyte of data were measured. From this initial data set, several data reduction steps will be performed to evolve a set of biomarkers which can best distinguish between the proteom of diseased and healthy patients.
We attempt to develop algorithms that apply the best machine-learning algorithms to this proteomics data: e.g., artificial neural network, supported vector machines, and multivariate analysis of discrimance.
The figure reveals a typical heatmap that was calculated from protein up- and downregulations in diseased and healthy subjects under different experimental conditions (e.g. wash conditions, chip surfaces etc.).

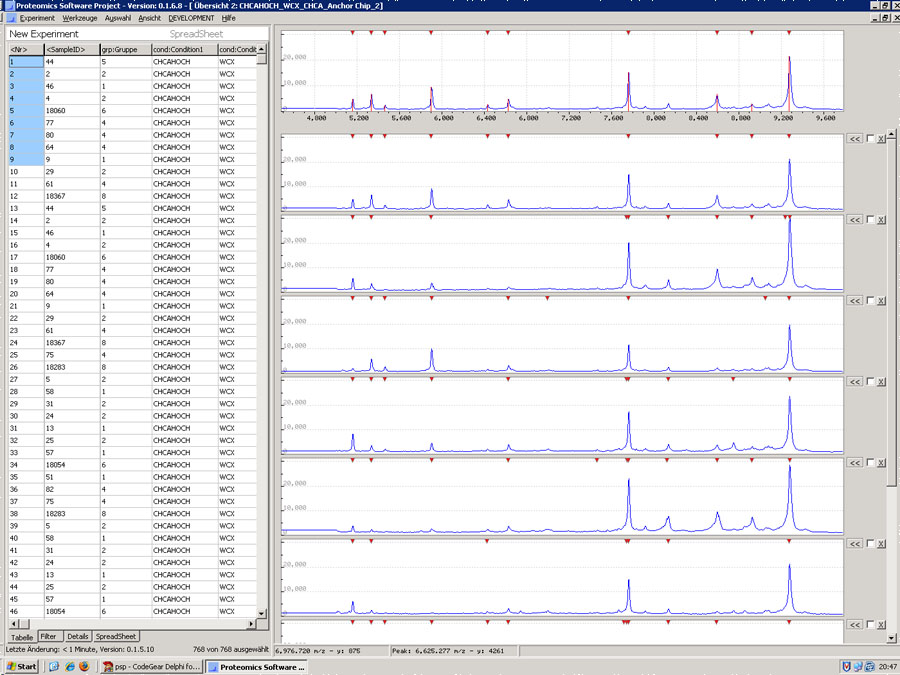
PSP – go fishing
PSP is a synonym for "Proteomics Software Project".
The aim of this project is to compare basically different mass spectrometry data from healthy and diseased samples derived from different mass spectrometers of different manufacturers e.g. Bruker and Biorad in their proprietary file formats.
At the moment the project is just prior to a first release candidate. The software will enable the user to search simultaneously huge amount of mass spectrometry data from different instruments for protein/peptide biomarkers by means of a variety of multivariate statistics, artificial neural networks and tree algorithms.
- Data Import from different Manufacturers
- Dynamic creation of study conditions and groups
- Displaying the spectra
- Comparing the spectra
- Spectra-Image Export
- Data Processing
- Spectra smoothing
- Baseline subtraction
- Normalisation
- Cluster data
- Create Matrix file for statistical analysis
- Interacting with Web-Server for statistical analysis
General Overview

General Overview with Peakdetection